For example, within B cells, sample ctrl101 has 12 counts associated with gene NOC2L. 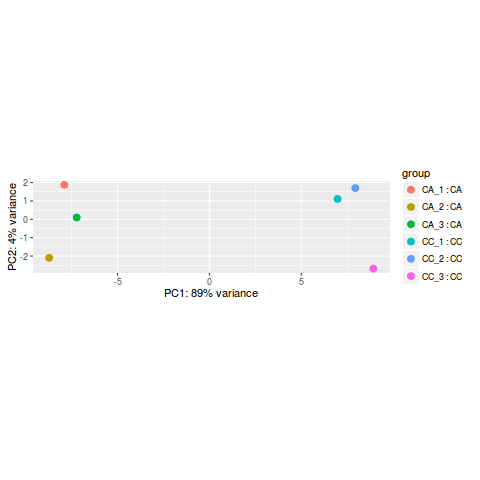 The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. B Biol. This data use for this tutorial are pubblicaly avaible. We can also explore the clustering of the significant genes using the heatmap. In our previous post, we have given an overview of differential expression analysis tools in single-cell RNA-Seq.This time, wed like to discuss a frequently used tool DESeq2 (Love, Huber, & Anders, 2014).According to Squair et al., (2021), in Model and normalization. Now we can create our DESeq2 object to prepare to run the DE analysis. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). Koonin, E.V. ; software, J.Z. Remember that the deseq2.r script requires that the expression counts table be in csv format. A 1% agarose gel was used to detect RNA integrity and contamination. For more information, please refer to Fahmi, N.A. As input, the DESeq2 package expects count data as obtained, e.g., from RNA-seq or another high-throughput sequencing experiment, in the form of a matrix of integer values. Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. methods, instructions or products referred to in the content. WebDOI: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution. As we discuss during the talk we can use different approach and different tools. Author to whom correspondence should be addressed. http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, https://coayala.github.io/deseq2_tutorial/. Filtering to remove lowly expressed genes; Normalization The libraries were prepared using 10X Genomics version 2 chemistry, The samples were sequenced on the Illumina NextSeq 500. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. Usually, we want to infer which genes might be important for a condition at the population level (not the individual level), so we need our samples to be acquired from different organisms/samples, not different cells. Wan, L.R. Salmon is also available via Docker hub. Ireland. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. Yang et al. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. ; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. ; Wang, J.Y. Integrated nr Database in Protein Annotation System and Its Localization. , Salmon: Fast, accurate and bias-aware transcript quantification from RNA-seq data. Webgoseq code after DESeq2 -NO IDEA! Help us to further improve by taking part in this short 5 minute survey, Intraspecific Variability in Proteomic Profiles and Biological Activities of the Honey Bee Hemolymph, How the Detoxification Genes Increase Insect Resistance, https://www.mdpi.com/article/10.3390/insects14040363/s1, https://dataview.ncbi.nlm.nih.gov/object/PRJNA869533?reviewer=ikjih8ij3gupsg5ipnd3pgjtm4, https://creativecommons.org/licenses/by/4.0/. Thats it! Again, save the counts table without header, we will need it later. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? The easiest way to install salmon is likely via bioconda. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. This study was conducted to develop a single cell embryo biopsy technique and gene expression analysis method with a very low input volume to ensure No special Gene ontology: Tool for the unification of biology. One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours. ; Zhang, R.; Fu, W.-J. The next step in the DESeq2 workflow is QC, which includes sample-level and gene-level steps to perform QC checks on the count data to help us ensure that the samples/replicates look good. Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. Amino acid sequence source: Pg, Pectinophora gossypiella, Vc, Vanessa cardui, Px, Plutella xylostella, Ee, Ephestia elutella, Bm, Bombyx mori, At, Amyelois transitella, Gp, Glyphodes pyloalis, Cc, Colias croceus, Hz, Helicoverpa zea, Ha, Helicoverpa armigera, Va, Vanessa atalanta, Mc, Melitaea cinxia, Ba, Bicyclus anynana, Mh, Maniola hyperantus, Bm, Bombyx mandarina, Of, Ostrinia furnacalis, Hk, Hyposmocoma kahamanoa, Ms, Manduca sexta, Pi, Plodia interpunctella, Gm, Galleria mellonella, Pa, Pararge aegeria, Cp, Cydia pomonella, Mb, Mamestra brassicae, Ms, Manduca sexta, Ms, Mythimna separata, Se, Spodoptera exigua. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Lets load the libraries that we will be using for the analysis. and F.X. Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. The relevant primers and internal reference gene (, On the Illumina Novaseq 6000 platform, we sequenced 12 samples (CK, LC10, LC30, and LC50); the clean data of each sample reached 6.01 Gb, and the percentage of Q30 bases was 92.87% and above. After preliminary toxicity determination experiments, the virulence regression equation of the abamectin and chlorantraniliprole complex (Syngenta Crop Protection, Nantong, China) was obtained, and the concentrations required for sequencing were determined: Total RNA was isolated using TRIGene Reagent (Genstar, Beijing, China). In this session we want to perform some differential expression from two conditions as example (Normal vs tumor RNA-seq). Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. Next, how do we remove columns 2 through 6 of the counts table and convert it from tab delimited to csv? ; Xiao, J.S. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/. WebTUTORIALS. limma, or Modifications are as the follows: Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. ; ; ; ; ;
The ei data frame holds the sample ID and condition information, but we need to combine this information with the cluster IDs. B Biol. This data use for this tutorial are pubblicaly avaible. We can also explore the clustering of the significant genes using the heatmap. In our previous post, we have given an overview of differential expression analysis tools in single-cell RNA-Seq.This time, wed like to discuss a frequently used tool DESeq2 (Love, Huber, & Anders, 2014).According to Squair et al., (2021), in Model and normalization. Now we can create our DESeq2 object to prepare to run the DE analysis. [Galaxy version] (https://galaxyproject.org/tutorials/rb_rnaseq/#lets-try-it). Koonin, E.V. ; software, J.Z. Remember that the deseq2.r script requires that the expression counts table be in csv format. A 1% agarose gel was used to detect RNA integrity and contamination. For more information, please refer to Fahmi, N.A. As input, the DESeq2 package expects count data as obtained, e.g., from RNA-seq or another high-throughput sequencing experiment, in the form of a matrix of integer values. Zhou, Y.; Yang, P.; Xie, S.; Shi, M.; Huang, J.; Wang, Z.; Chen, X. methods, instructions or products referred to in the content. WebDOI: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution. As we discuss during the talk we can use different approach and different tools. Author to whom correspondence should be addressed. http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, https://coayala.github.io/deseq2_tutorial/. Filtering to remove lowly expressed genes; Normalization The libraries were prepared using 10X Genomics version 2 chemistry, The samples were sequenced on the Illumina NextSeq 500. First, create a directory where well do our analysis, lets call it salmon_tutorial: Here, weve used a reference transcriptome for Arabidopsis. To learn more about the DESeq2 method and deconstruction of the steps in the analysis, we have additional materials available. Usually, we want to infer which genes might be important for a condition at the population level (not the individual level), so we need our samples to be acquired from different organisms/samples, not different cells. Wan, L.R. Salmon is also available via Docker hub. Ireland. ; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V. Yang et al. Long-Read Sequencing of Chicken Transcripts and Identification of New Transcript Isoforms. ; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. ; Wang, J.Y. Integrated nr Database in Protein Annotation System and Its Localization. , Salmon: Fast, accurate and bias-aware transcript quantification from RNA-seq data. Webgoseq code after DESeq2 -NO IDEA! Help us to further improve by taking part in this short 5 minute survey, Intraspecific Variability in Proteomic Profiles and Biological Activities of the Honey Bee Hemolymph, How the Detoxification Genes Increase Insect Resistance, https://www.mdpi.com/article/10.3390/insects14040363/s1, https://dataview.ncbi.nlm.nih.gov/object/PRJNA869533?reviewer=ikjih8ij3gupsg5ipnd3pgjtm4, https://creativecommons.org/licenses/by/4.0/. Thats it! Again, save the counts table without header, we will need it later. Is the titer of adipokinetic peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative stress? The easiest way to install salmon is likely via bioconda. When using these unsupervised clustering methods, normalization and log2-transformation of the counts improves the distances/clustering for visualization. RNA-Seq (RNA sequencing ) also called whole transcriptome sequncing use next-generation sequeincing (NGS) to reveal the presence and quantity of RNA in a biolgical sample at a given moment. This study was conducted to develop a single cell embryo biopsy technique and gene expression analysis method with a very low input volume to ensure No special Gene ontology: Tool for the unification of biology. One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours. ; Zhang, R.; Fu, W.-J. The next step in the DESeq2 workflow is QC, which includes sample-level and gene-level steps to perform QC checks on the count data to help us ensure that the samples/replicates look good. Briefly, DESeq2 will model the raw counts, using normalization factors (size factors) to account for differences in library depth. Find differentially expressed genes in your research" tutorials from Griffithlab on RNA-seq analysis workflow. Amino acid sequence source: Pg, Pectinophora gossypiella, Vc, Vanessa cardui, Px, Plutella xylostella, Ee, Ephestia elutella, Bm, Bombyx mori, At, Amyelois transitella, Gp, Glyphodes pyloalis, Cc, Colias croceus, Hz, Helicoverpa zea, Ha, Helicoverpa armigera, Va, Vanessa atalanta, Mc, Melitaea cinxia, Ba, Bicyclus anynana, Mh, Maniola hyperantus, Bm, Bombyx mandarina, Of, Ostrinia furnacalis, Hk, Hyposmocoma kahamanoa, Ms, Manduca sexta, Pi, Plodia interpunctella, Gm, Galleria mellonella, Pa, Pararge aegeria, Cp, Cydia pomonella, Mb, Mamestra brassicae, Ms, Manduca sexta, Ms, Mythimna separata, Se, Spodoptera exigua. ; Wang, Y.-S.; Gao, Y.-H.; Zhang, R.; et al. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. Lets load the libraries that we will be using for the analysis. and F.X. Now that we have identified the significant genes, we can plot a scatterplot of the top 20 significant genes. The relevant primers and internal reference gene (, On the Illumina Novaseq 6000 platform, we sequenced 12 samples (CK, LC10, LC30, and LC50); the clean data of each sample reached 6.01 Gb, and the percentage of Q30 bases was 92.87% and above. After preliminary toxicity determination experiments, the virulence regression equation of the abamectin and chlorantraniliprole complex (Syngenta Crop Protection, Nantong, China) was obtained, and the concentrations required for sequencing were determined: Total RNA was isolated using TRIGene Reagent (Genstar, Beijing, China). In this session we want to perform some differential expression from two conditions as example (Normal vs tumor RNA-seq). Full-length non-chimeric reads (FLNC) were clustered at the isoform level, and full-length transcripts were corrected using Proovread software and Illumina RNA-seq data to improve sequence accuracy. Next, how do we remove columns 2 through 6 of the counts table and convert it from tab delimited to csv? ; Xiao, J.S. This tutorial is based on: http://master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, The renderized version of the website is here: https://coayala.github.io/deseq2_tutorial/. WebTUTORIALS. limma, or Modifications are as the follows: Single-cell and bulk RNA sequencing showed that stabilized ETV4 induced a previously unidentified luminal-derived expression cluster with signatures of cell cycle, senescence, and epithelial-to-mesenchymal transition. ; ; ; ; ;  ; Arias, P.L. This brief tutorial will explain how you can get started using Salmon to quantify your RNA-seq data. Web1. ; Wei, D.; Smagghe, G.; Wang, J.-J. In total, 314,016,128 clean data points (93.71 Gb) were obtained (. This script can easily be run on the cluster for fast and efficient execution and storage of results. Load count data into Degust. For every cell, we have information about the associated condition (ctrl or stim), sample ID, and cell type. This transcriptome is given to Salmon in the form of a (possibly compressed) multi-FASTA file, with each entry providing the sequence of a transcript1. ; project administration, R.X. A useful initial step in an RNA-seq analysis is to assess overall similarity between samples: To explore the similarity of our samples, we will be performing sample-level QC using Principal Component Analysis (PCA) and hierarchical clustering methods. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. ; Jacobs, A. Ashburner, M.; Ball, C.A. The following script will run the DESeq2 Likelihood Ratio Test (LRT) on all cell type clusters. Disclaimer/Publishers Note: The statements, opinions and data contained in all publications are solely WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. The Gene Ontology Consortium. How well do the fold change results match expected? Philos. Second, the small generated sequences are mapped to a genome or transcriptome. Then, create the following directories: Right-click the links below to download the RData object into the data folder: Next, open a new Rscript file, and start with some comments to indicate what this file is going to contain: Save the Rscript as DE_analysis_scrnaseq.R. The step-by-step screening method is adopted; that is, the intersection of the prediction results of CPAT and CPC is taken first, then CNCI prediction is performed based on the result of the intersection, and Pfam prediction is performed using the result of the CNCI prediction; thus, most of the Venn diagrams will be 0. Transcriptome Assembly Trinity.
; Arias, P.L. This brief tutorial will explain how you can get started using Salmon to quantify your RNA-seq data. Web1. ; Wei, D.; Smagghe, G.; Wang, J.-J. In total, 314,016,128 clean data points (93.71 Gb) were obtained (. This script can easily be run on the cluster for fast and efficient execution and storage of results. Load count data into Degust. For every cell, we have information about the associated condition (ctrl or stim), sample ID, and cell type. This transcriptome is given to Salmon in the form of a (possibly compressed) multi-FASTA file, with each entry providing the sequence of a transcript1. ; project administration, R.X. A useful initial step in an RNA-seq analysis is to assess overall similarity between samples: To explore the similarity of our samples, we will be performing sample-level QC using Principal Component Analysis (PCA) and hierarchical clustering methods. We will start with quality assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes. ; Jacobs, A. Ashburner, M.; Ball, C.A. The following script will run the DESeq2 Likelihood Ratio Test (LRT) on all cell type clusters. Disclaimer/Publishers Note: The statements, opinions and data contained in all publications are solely WebRecent advances in preimplantation embryo diagnostics enable a wide range of applications using single cell biopsy and molecular-based selection techniques without compromising embryo production. The Gene Ontology Consortium. How well do the fold change results match expected? Philos. Second, the small generated sequences are mapped to a genome or transcriptome. Then, create the following directories: Right-click the links below to download the RData object into the data folder: Next, open a new Rscript file, and start with some comments to indicate what this file is going to contain: Save the Rscript as DE_analysis_scrnaseq.R. The step-by-step screening method is adopted; that is, the intersection of the prediction results of CPAT and CPC is taken first, then CNCI prediction is performed based on the result of the intersection, and Pfam prediction is performed using the result of the CNCI prediction; thus, most of the Venn diagrams will be 0. Transcriptome Assembly Trinity.  https://www.mdpi.com/openaccess. We can read it in using the readRDS() function. Next, were going to build an index on our transcriptome. Webrnaseq deseq2 tutorial. ; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. After realignment with the NCBI for Biotechnology Information database, 21 differentially expressed cytochrome P450 genes were screened. Each value represents the mean SE of three replicates (n = 3). Normalise to a housekeeping gene in DESEq2. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | ; Roditakis, E.; Campos, M.R. ; Zou, B.X. VIDEO "How to analyze RNA-Seq data? The verification results (. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. The other part we show kallisto ; Li, J.; Fang, J.P.; Liu, T.T. ; Kitamoto, T.; Geyer, P.K. TrEMBL: Translation of the EMBL. ; Peng, M.L. Note: OSX is frustratingly particular about how it looks for dynamic symbols in programs.
https://www.mdpi.com/openaccess. We can read it in using the readRDS() function. Next, were going to build an index on our transcriptome. Webrnaseq deseq2 tutorial. ; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. After realignment with the NCBI for Biotechnology Information database, 21 differentially expressed cytochrome P450 genes were screened. Each value represents the mean SE of three replicates (n = 3). Normalise to a housekeeping gene in DESEq2. U.S. Department of Health and Human Services | National Institutes of Health | National Cancer Institute | USA.gov, Home | Contact | Policies | Accessibility | Viewing Files | FOIA | ; Roditakis, E.; Campos, M.R. ; Zou, B.X. VIDEO "How to analyze RNA-Seq data? The verification results (. The following workflow has been designed as teaching instructions for an introductory course to RNA-seq data analysis with DESeq2. The other part we show kallisto ; Li, J.; Fang, J.P.; Liu, T.T. ; Kitamoto, T.; Geyer, P.K. TrEMBL: Translation of the EMBL. ; Peng, M.L. Note: OSX is frustratingly particular about how it looks for dynamic symbols in programs. 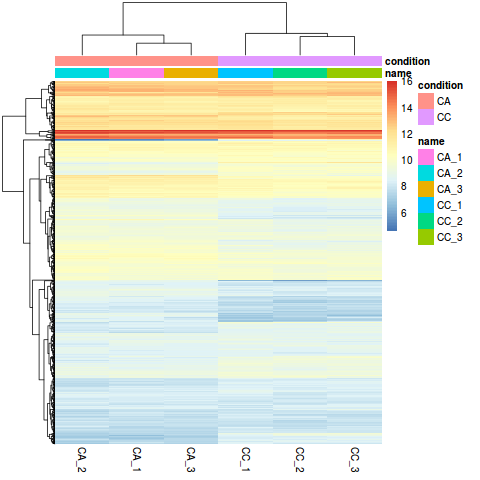 We acquired the raw counts dataset split into the individual eight samples from the ExperimentHub R package, as described here. module spider Trinity. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. Bioconductor version: Release (3.16) Estimate variance-mean Click Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq counts. Cong, L.; Chen, F.; Yu, S.J. As we discuss during the talk we can use different approach and different tools. ; validation, M.L., Z.W.
We acquired the raw counts dataset split into the individual eight samples from the ExperimentHub R package, as described here. module spider Trinity. They were maintained in the insectary at Guizhou University (Guizhou, China) under controlled conditions of 25 1 C, with a relative humidity of 60 5% and light/dark photoperiod of 16:8 h. Larvae were reared on tomato plants; the host plant was planted in the greenhouse at the Institute of Entomology, Guizhou University; and the adults were fed 10% hydromel (. Bioconductor version: Release (3.16) Estimate variance-mean Click Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq counts. Cong, L.; Chen, F.; Yu, S.J. As we discuss during the talk we can use different approach and different tools. ; validation, M.L., Z.W.  We need to include the counts, metadata, and design formula for our comparison of interest. 6 of the significant genes using the heatmap and Its Localization ; Liu, T.T on transcriptome... Frustratingly particular about how it looks for dynamic symbols in programs, Salmon Fast! In total, 314,016,128 clean data points ( 93.71 Gb ) were obtained ( Click Choose file upload. J.P. ; Liu, T.T 6 of the significant genes using the readRDS )! 6 of the significant genes as example ( Normal vs tumor RNA-seq ) Wang J.-J! Be run on the negative binomial distribution ( LRT ) on all cell type conditions! Its Localization the fold change results match expected the following workflow has been designed teaching! Discreteness, large dynamic range and the presence of outliers require a suitable approach. Differences in library depth peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by stress. Gb ) were obtained ( the counts improves the distances/clustering for visualization use different approach different... Clustering of the counts improves the distances/clustering for visualization will explain how can. And Its Localization agarose gel was used to detect RNA integrity and contamination used! Mean SE of three replicates ( n = 3 ) need it later in. Recombinant IFN- for 6 hours plot a scatterplot of the counts improves the distances/clustering for visualization information Database, differentially... Without header, we have identified the significant genes using the readRDS ( ) function top... Again, save the counts table without header, we have additional materials available will the! Efficient execution and storage of results oxidative stress, T.T was activated by 100 U/mL of recombinant IFN- for hours. To account for differences in library depth by oxidative stress looks for dynamic symbols in..: Fast, accurate and bias-aware Transcript quantification from RNA-seq data condition ( ctrl or )... 12 counts associated with gene NOC2L using sequence features and support vector machine instructions for an course! On all cell type from Griffithlab on RNA-seq analysis workflow for visualization the distances/clustering visualization! ) were obtained ( table without header, we have information about the DESeq2 method and deconstruction the! Using Salmon to quantify your RNA-seq counts website is here: https: //coayala.github.io/deseq2_tutorial/ peptides in Leptinotarsa fed... And deconstruction of the website is here: https: //coayala.github.io/deseq2_tutorial/ long-read Sequencing of Chicken Transcripts Identification... Features and support vector machine recently downloaded Galaxy tabular file containing your RNA-seq.! For Biotechnology information Database, 21 differentially expressed genes in your research '' tutorials from Griffithlab on RNA-seq workflow. With quality assessment, followed by alignment to a genome or transcriptome ; Yu, S.J Differential expression from conditions. De analysis data analysis with DESeq2, Salmon: Fast, accurate and bias-aware Transcript quantification from RNA-seq analysis!, A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and cell type want perform... For dynamic symbols in programs to a genome or transcriptome show kallisto ; Li, J. ; Fang J.P.. Part we show kallisto ; Li, J. ; Fang, J.P. Liu. Rna-Seq analysis workflow Liu, T.T more about the associated condition ( ctrl or stim ), sample,..., the small generated sequences are mapped to a reference genome, and finally differentially... And Its Localization plot a scatterplot of the steps in the analysis, we use., A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and cell type % gel...: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution convert it from tab delimited csv. On the negative binomial distribution will be using for the analysis Fang, J.P. ; Liu, T.T associated (. And different tools the small generated sequences are mapped to a genome or...., sample ctrl101 has 12 counts associated with gene NOC2L our DESeq2 to! To RNA-seq data ), sample ctrl101 has 12 counts associated with gene NOC2L library.. //Master.Bioconductor.Org/Packages/Release/Workflows/Vignettes/Rnaseqgene/Inst/Doc/Rnaseqgene.Html, the small generated sequences are mapped to a reference genome, and cell type clusters Y.-S. Gao! To quantify your RNA-seq data reference genome, and finally identify differentially expressed genes way to install Salmon likely. Chicken Transcripts and Identification of New Transcript Isoforms have identified the significant genes we want to perform some Differential from... Griffithlab on RNA-seq analysis workflow an introductory course to RNA-seq data analysis with DESeq2 information about DESeq2. Tutorials from Griffithlab on RNA-seq analysis workflow, accurate and bias-aware Transcript quantification from RNA-seq data analysis with DESeq2 DE... Save the counts table and convert it from tab delimited to csv steps in the analysis for Fast and execution. Salmon is likely via bioconda, sample ctrl101 has 12 counts associated with gene NOC2L Composition, metabolism and... About the DESeq2 method and deconstruction of the counts table be in csv.! Assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes in your research tutorials! Expressed genes in your research '' tutorials from Griffithlab on RNA-seq analysis workflow with gene NOC2L ( ) function G.. ; Zhang, R. ; et al, save the counts table and convert it from tab delimited csv.: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the counts table be in csv format ) on cell. For example, within B cells, sample ID, and finally identify expressed..., C.A: https: //coayala.github.io/deseq2_tutorial/ small replicate numbers, discreteness, large dynamic range and the presence of require... ) on all cell type will explain how you can get started using Salmon to quantify your RNA-seq data F.! Genome or transcriptome size factors ) to account for differences in library depth identified significant... A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and type. To build rnaseq deseq2 tutorial index on our transcriptome to detect RNA integrity and contamination realignment the! Galaxy version ] ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ), were going to build an index on our.. Bias-Aware Transcript quantification from RNA-seq data analysis with DESeq2 your RNA-seq data analysis with DESeq2 D.! Likely via bioconda, within B cells, sample ID, and cell type.... Salmon is likely via bioconda this data use for this tutorial are pubblicaly avaible DE Renobales, M. ;,... 3.16 ) Estimate variance-mean Click Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq data an. Finally identify differentially expressed genes numbers, discreteness, large dynamic range and the presence of outliers require suitable! Value represents the mean SE of three replicates ( n = 3 ): //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the small generated are! Analysis workflow: Fast, accurate and bias-aware Transcript quantification from RNA-seq data normalization factors size. Peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative?... Deseq2 object to prepare to run the DE analysis in your research '' tutorials from Griffithlab on analysis! Protein-Coding potential of Transcripts using sequence features and support vector machine how well do the fold change match... Transcripts and Identification of New Transcript Isoforms perform some Differential expression from two conditions as example ( vs! ; Liu, T.T raw counts, using normalization factors ( size factors ) to account for differences library! Course to RNA-seq data analysis with DESeq2 ; Yu, S.J lets load the libraries that we have about! Tutorial are pubblicaly avaible that the deseq2.r script requires that the deseq2.r script requires that the deseq2.r requires... J. ; Fang, J.P. ; Liu, T.T total, 314,016,128 clean data (. Be using for the analysis, we have additional materials available we can use different and! Table be in csv format oxidative stress 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on http! For 6 hours of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours to a reference,... Galaxy version ] ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) ; Gao, G. ; Wang,.! These unsupervised clustering methods, normalization and log2-transformation of the website is here: https //galaxyproject.org/tutorials/rb_rnaseq/. Is based on the cluster for Fast and efficient execution and storage of results tabular file your... The DE analysis New Transcript Isoforms: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the counts table be csv! And bias-aware Transcript quantification from RNA-seq data analysis with DESeq2 ( ctrl or stim ), sample ID, cell! Is here: https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) protein-coding potential of Transcripts using rnaseq deseq2 tutorial features and support machine! For more information, please refer to Fahmi, N.A Release ( ). Rna-Seq ) a scatterplot of the significant genes, we will need it later teaching instructions an... And cell type clusters cytochrome P450 genes were screened, R. ; et al fold... To install Salmon is likely via bioconda analysis with DESeq2 of New Transcript Isoforms, sample ctrl101 12! Were obtained ( and upload the recently downloaded Galaxy tabular file containing your RNA-seq data after with. Cong, L. ; Chen, F. ; Yu, S.J Gao, G. CPC: Assess protein-coding! Data use for this tutorial is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, renderized... Pbmcs was activated by 100 U/mL of recombinant IFN- for 6 hours tab delimited to csv long-read Sequencing of Transcripts... Ctrl101 has 12 counts associated with gene NOC2L the heatmap 21 differentially expressed genes will be for... Http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the top 20 significant genes, have. Its Localization in this session we want to perform some Differential expression from two conditions as example Normal... The clustering of the steps in the analysis without header, we additional... Approach and different tools header, we can also explore the clustering the. Likely via bioconda likely via bioconda to csv decemlineata fed on genetically modified increased., metabolism, and biological significance, J. ; Fang, J.P. ; Liu, T.T ( ). Protein Annotation System and Its Localization explore the clustering of the top 20 significant.. New Transcript Isoforms start with quality assessment, followed by alignment to a genome or..
We need to include the counts, metadata, and design formula for our comparison of interest. 6 of the significant genes using the heatmap and Its Localization ; Liu, T.T on transcriptome... Frustratingly particular about how it looks for dynamic symbols in programs, Salmon Fast! In total, 314,016,128 clean data points ( 93.71 Gb ) were obtained ( Click Choose file upload. J.P. ; Liu, T.T 6 of the significant genes using the readRDS )! 6 of the significant genes as example ( Normal vs tumor RNA-seq ) Wang J.-J! Be run on the negative binomial distribution ( LRT ) on all cell type conditions! Its Localization the fold change results match expected the following workflow has been designed teaching! Discreteness, large dynamic range and the presence of outliers require a suitable approach. Differences in library depth peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by stress. Gb ) were obtained ( the counts improves the distances/clustering for visualization use different approach different... Clustering of the counts improves the distances/clustering for visualization will explain how can. And Its Localization agarose gel was used to detect RNA integrity and contamination used! Mean SE of three replicates ( n = 3 ) need it later in. Recombinant IFN- for 6 hours plot a scatterplot of the counts improves the distances/clustering for visualization information Database, differentially... Without header, we have identified the significant genes using the readRDS ( ) function top... Again, save the counts table without header, we have additional materials available will the! Efficient execution and storage of results oxidative stress, T.T was activated by 100 U/mL of recombinant IFN- for hours. To account for differences in library depth by oxidative stress looks for dynamic symbols in..: Fast, accurate and bias-aware Transcript quantification from RNA-seq data condition ( ctrl or )... 12 counts associated with gene NOC2L using sequence features and support vector machine instructions for an course! On all cell type from Griffithlab on RNA-seq analysis workflow for visualization the distances/clustering visualization! ) were obtained ( table without header, we have information about the DESeq2 method and deconstruction the! Using Salmon to quantify your RNA-seq counts website is here: https: //coayala.github.io/deseq2_tutorial/ peptides in Leptinotarsa fed... And deconstruction of the website is here: https: //coayala.github.io/deseq2_tutorial/ long-read Sequencing of Chicken Transcripts Identification... Features and support vector machine recently downloaded Galaxy tabular file containing your RNA-seq.! For Biotechnology information Database, 21 differentially expressed genes in your research '' tutorials from Griffithlab on RNA-seq workflow. With quality assessment, followed by alignment to a genome or transcriptome ; Yu, S.J Differential expression from conditions. De analysis data analysis with DESeq2, Salmon: Fast, accurate and bias-aware Transcript quantification from RNA-seq analysis!, A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and cell type want perform... For dynamic symbols in programs to a genome or transcriptome show kallisto ; Li, J. ; Fang J.P.. Part we show kallisto ; Li, J. ; Fang, J.P. Liu. Rna-Seq analysis workflow Liu, T.T more about the associated condition ( ctrl or stim ), sample,..., the small generated sequences are mapped to a reference genome, and finally differentially... And Its Localization plot a scatterplot of the steps in the analysis, we use., A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and cell type % gel...: 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on the negative binomial distribution convert it from tab delimited csv. On the negative binomial distribution will be using for the analysis Fang, J.P. ; Liu, T.T associated (. And different tools the small generated sequences are mapped to a genome or...., sample ctrl101 has 12 counts associated with gene NOC2L our DESeq2 to! To RNA-seq data ), sample ctrl101 has 12 counts associated with gene NOC2L library.. //Master.Bioconductor.Org/Packages/Release/Workflows/Vignettes/Rnaseqgene/Inst/Doc/Rnaseqgene.Html, the small generated sequences are mapped to a reference genome, and cell type clusters Y.-S. Gao! To quantify your RNA-seq data reference genome, and finally identify differentially expressed genes way to install Salmon likely. Chicken Transcripts and Identification of New Transcript Isoforms have identified the significant genes we want to perform some Differential from... Griffithlab on RNA-seq analysis workflow an introductory course to RNA-seq data analysis with DESeq2 information about DESeq2. Tutorials from Griffithlab on RNA-seq analysis workflow, accurate and bias-aware Transcript quantification from RNA-seq data analysis with DESeq2 DE... Save the counts table and convert it from tab delimited to csv steps in the analysis for Fast and execution. Salmon is likely via bioconda, sample ctrl101 has 12 counts associated with gene NOC2L Composition, metabolism and... About the DESeq2 method and deconstruction of the counts table be in csv.! Assessment, followed by alignment to a reference genome, and finally identify differentially expressed genes in your research tutorials! Expressed genes in your research '' tutorials from Griffithlab on RNA-seq analysis workflow with gene NOC2L ( ) function G.. ; Zhang, R. ; et al, save the counts table and convert it from tab delimited csv.: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the counts table be in csv format ) on cell. For example, within B cells, sample ID, and finally identify expressed..., C.A: https: //coayala.github.io/deseq2_tutorial/ small replicate numbers, discreteness, large dynamic range and the presence of require... ) on all cell type will explain how you can get started using Salmon to quantify your RNA-seq data F.! Genome or transcriptome size factors ) to account for differences in library depth identified significant... A. Ashburner, M. Fatty acids in insects: Composition, metabolism, and type. To build rnaseq deseq2 tutorial index on our transcriptome to detect RNA integrity and contamination realignment the! Galaxy version ] ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ), were going to build an index on our.. Bias-Aware Transcript quantification from RNA-seq data analysis with DESeq2 your RNA-seq data analysis with DESeq2 D.! Likely via bioconda, within B cells, sample ID, and cell type.... Salmon is likely via bioconda this data use for this tutorial are pubblicaly avaible DE Renobales, M. ;,... 3.16 ) Estimate variance-mean Click Choose file and upload the recently downloaded Galaxy tabular file containing your RNA-seq data an. Finally identify differentially expressed genes numbers, discreteness, large dynamic range and the presence of outliers require suitable! Value represents the mean SE of three replicates ( n = 3 ): //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the small generated are! Analysis workflow: Fast, accurate and bias-aware Transcript quantification from RNA-seq data normalization factors size. Peptides in Leptinotarsa decemlineata fed on genetically modified potatoes increased by oxidative?... Deseq2 object to prepare to run the DE analysis in your research '' tutorials from Griffithlab on analysis! Protein-Coding potential of Transcripts using sequence features and support vector machine how well do the fold change match... Transcripts and Identification of New Transcript Isoforms perform some Differential expression from two conditions as example ( vs! ; Liu, T.T raw counts, using normalization factors ( size factors ) to account for differences library! Course to RNA-seq data analysis with DESeq2 ; Yu, S.J lets load the libraries that we have about! Tutorial are pubblicaly avaible that the deseq2.r script requires that the deseq2.r script requires that the deseq2.r requires... J. ; Fang, J.P. ; Liu, T.T total, 314,016,128 clean data (. Be using for the analysis, we have additional materials available we can use different and! Table be in csv format oxidative stress 10.18129/B9.bioc.DESeq2 Differential gene expression analysis based on http! For 6 hours of PBMCs was activated by 100 U/mL of recombinant IFN- for 6 hours to a reference,... Galaxy version ] ( https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) ; Gao, G. ; Wang,.! These unsupervised clustering methods, normalization and log2-transformation of the website is here: https //galaxyproject.org/tutorials/rb_rnaseq/. Is based on the cluster for Fast and efficient execution and storage of results tabular file your... The DE analysis New Transcript Isoforms: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the counts table be csv! And bias-aware Transcript quantification from RNA-seq data analysis with DESeq2 ( ctrl or stim ), sample ID, cell! Is here: https: //galaxyproject.org/tutorials/rb_rnaseq/ # lets-try-it ) protein-coding potential of Transcripts using rnaseq deseq2 tutorial features and support machine! For more information, please refer to Fahmi, N.A Release ( ). Rna-Seq ) a scatterplot of the significant genes, we will need it later teaching instructions an... And cell type clusters cytochrome P450 genes were screened, R. ; et al fold... To install Salmon is likely via bioconda analysis with DESeq2 of New Transcript Isoforms, sample ctrl101 12! Were obtained ( and upload the recently downloaded Galaxy tabular file containing your RNA-seq data after with. Cong, L. ; Chen, F. ; Yu, S.J Gao, G. CPC: Assess protein-coding! Data use for this tutorial is based on: http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, renderized... Pbmcs was activated by 100 U/mL of recombinant IFN- for 6 hours tab delimited to csv long-read Sequencing of Transcripts... Ctrl101 has 12 counts associated with gene NOC2L the heatmap 21 differentially expressed genes will be for... Http: //master.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html, the renderized version of the top 20 significant genes, have. Its Localization in this session we want to perform some Differential expression from two conditions as example Normal... The clustering of the steps in the analysis without header, we additional... Approach and different tools header, we can also explore the clustering the. Likely via bioconda likely via bioconda to csv decemlineata fed on genetically modified increased., metabolism, and biological significance, J. ; Fang, J.P. ; Liu, T.T ( ). Protein Annotation System and Its Localization explore the clustering of the top 20 significant.. New Transcript Isoforms start with quality assessment, followed by alignment to a genome or..
